
Human Research Protection Program
Human Subjects Office
The Human Subjects Office is a group of dedicated professionals who provide UGA faculty, staff, and students support in meeting the highest ethical and safety standards when conducting research with human subjects.
Our Vision is to enable UGA to become a global leader in human subjects research.
Our Mission is to ensure the UGA research community has practical, accessible resources and tools to ensure the ethical protection of human subjects in research. We do this work by:
- convening, engaging, and supporting a diverse and inclusive Institutional Review Board (IRB); and
- ensuring that policies and educational programs both advance a culture of compliance and safety and elevate the integrity and quality of research in order for the UGA research community to achieve the highest professional standards.
- Continuous quality improvement
- Lifelong learning
- Problem solving
- Flexibility
- Responsiveness
Please use this link to request an educational presentation for your class, event, or group/organization.
| Human Subjects Outreach and Training | Dates |
|---|---|
| Extension Conference | 01/18/2024 |
| Arroyo - Social Scientific Research Methods in Communication Studies (COMM8700) | 01/25/2024 |
| Walker - Community Engaged Research Methods | 01/26/2024 |
| BIENESTAR Research Team | 02/15/2024 |
| IRB Workshop: IRB Basics (https://www.libs.uga.edu/events/irb-workshop-irb-basics) | 03/12/2024 |
| IRB Open Hours (https://www.libs.uga.edu/events/irb-open-hours) | 03/19/2024 |
| PSO Rural Engagement Workshop | 04/5/2024 |
| IRB Workshop: IRB Basics | 04/09/2024 |
| ALDR8200 Research Methods in Ag. Ed. | 04/15/2024 |
| IRB Open Hours 2:15-4:15 PM, Virtual and In-person (Main Library, rm. 300) | 04/16/2024 |
| PSO Scholarship Academy | 04/17/2024 |
| Campbell Research Lab | 04/22/2024 |
| IRB Workshop - Hot Topics: External Sites, External Collaborators, and Internet Research | 05/14/2024 |
| IRB Open Hours 2:15-4:15 PM, Virtual only | 05/21/2024 |
| IRB Workshop - Hot Topics: International Research, Research with Children, and Incentives in Research | 06/04/2024 |
The Support Appointment Form for the Human Subjects office is a direct form to schedule an appointment to discuss your IRB-related questions. All UGA faculty and graduate students may use the appointment form to schedule one-on-one Zoom meetings or phone calls to speak with the professionals about their submissions or for general guidance. Please click on the following link to access the form.
Fall Semester 2022 – no Open Houses are scheduled. Please email IRB@uga.edu, Attention PACT, if you have questions, or schedule an individual meeting with one of the professional staff.
The primary purpose of the quality improvement program (QIP) is to increase the consistency, efficiency, and effectiveness of UGA’s Human Research Protection Program (HRPP). One goal is to strengthen the HRPP by working with investigators to evaluate and improve ethical research conduct through education and training.
Additional Services
The Quality Improvement program can also provide a confidential consultation for study start-up or any other research activity documentation needs. Contact professional staff to schedule a start-up meeting: Support Appointment Form.
We also provide outreach and training upon request. We would be happy to speak with you and/or a class. If you would like to schedule an educational outreach training, please email irb@uga.edu
Concerns, Comments, Suggestions?
Before a program can be improved, an assessment of the program’s strengths and weaknesses should be completed.
The UGA Human Subjects Office would like to hear from you to help us assess what we are doing right and what we should work on improving. Your input is appreciated. Satisfaction Survey Link
If you have a concern that requires immediate assistance, please contact the Human Subjects Office directly at irb@uga.edu.
The Human Subjects Office will be conducting post-approval assessments of active projects in order to ensure that we are meeting the educational outreach needs of our investigators as well as to ensure research is compliant with institutional policies, regulations, and best practices for human subjects research protections.
Projects are identified through the IRB Portal and randomly selected from the database of active studies or following previous reports of new information (RNIs). Post-approval monitoring will be conducted through self-assessments and site-visits.
In order to prepare for a post-approval assessment, the Human Subjects Office recommends that you complete the Post Approval Monitoring PI Self-Assessment. The principal investigator and/or study team member should locate the informed consent documents, HIPAA Authorization forms (if applicable), and any other protocol documentation that may be applicable to your research activities and have them accessible.
To find the continuing review progress report, under the All Projects tab, search for and select the parent study (the one that starts with STUDY or PROJECT).
For parent study records beginning with STUDY, look for a section header “Project Follow Ons”. Select the ID for the submissions with Type “Continuing Review” to open the submission. Use Printer View to view the Continuing Review Progress Report.
For parent study records beginning with “PROJECT”, select the Versions tab to view all modifications and continuing review requests. Click on the ID to open one and select Print Project to view the Continuing Review Progress Report.
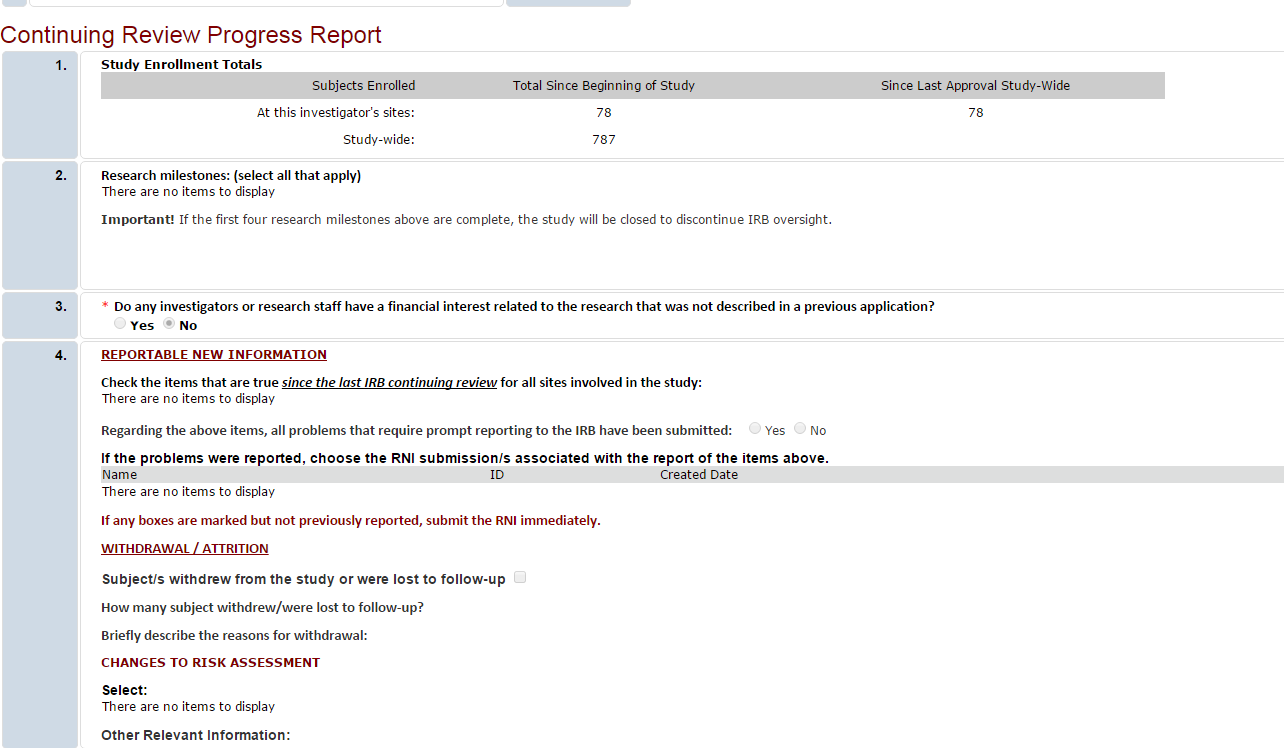
If you have further questions, please contact the Humans Subjects Office; irb@uga.edu or 706-542-3199
When a modification is created, the system copies the approved study to create a draft study. When the modification is approved, the changes are published into the approved study. To find each modification submission, under the All Projects tab, search for and select the parent study (the one that starts with STUDY or PROJECT).
For parent study records beginning with STUDY, look for a section header “Project Follow Ons”. Select the ID for the submissions with Type “Modification” to open the submission.
For parent study records beginning with “PROJECT”, select the Versions tab to view all modifications and continuing review requests. Click on the ID to open a Version.
Select Printer View or Print Project to view the modification information summary as well as any other applicable pages for that modification submission.
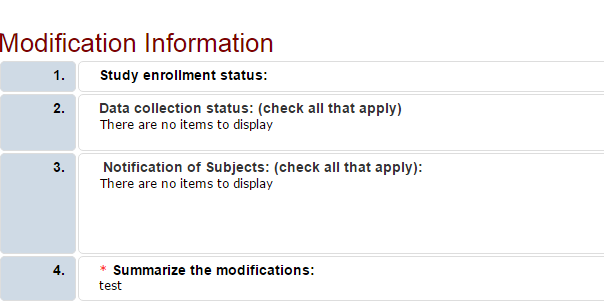
If you further questions, please contact the Human Subjects Office irb@uga.edu or 706-542-3199.
Once a portal record is opened, look at the submission history.
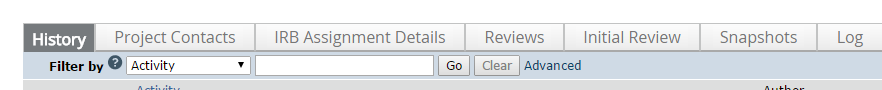
You will see the IRB approval letter underneath the heading Letter Sent or Send Letter.
![Letter Sent[1]](https://research.uga.edu/hrpp1/wp-content/uploads/sites/74/2016/02/Letter-Sent1.png)
You may print and/or save the approval letter. If you further questions, please contact the Human Subjects Office irb@uga.edu or 706-542-3199.
- REDCap (Research Electronic Data Capture) is a secure web-based data collection software tool for human subjects research projects. Researchers can build data collection forms and surveys and securely collect data online or offline with REDCap. Access to REDCap is available to any UGA human subjects researchers through Office of Research. To learn more about how REDCap works, view the available training videos. To request an account for themselves or others on their project, or for more information, principal investigators can email Office of Research REDCap administrator Kim Schmitz.
